 |
|
Case Report
| ||||||
| Primary ovarian insufficiency in a xeroderma pigmentosum patient with consanguineous parents | ||||||
| Abdullah K. Agabawi1, Wafa M. Faqeeh2, Amal A. Kokandi3, Hisham Nassief1 | ||||||
|
1Demonstrator, Obstetrics and Gynecology Department, King Abdulaziz University Hospital, Jeddah, Saudi Arabia 2Professor, Obstetrics and Gynecology department, King Abdulaziz University Hospital, Jeddah, Saudi Arabia 3Assistant Professor, Department of Medicine, Dermatology Unit, King Abdulaziz University Hospital, Jeddah, Saudi Arabia | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] |


| How to cite this article |
| Agabawi AK, Faqeeh WM, Kokandi AA, Nassief H. Primary ovarian insufficiency in a xeroderma pigmentosum patient with consanguineous parents. J Case Rep Images Obstet Gynecol 2018;4:1–7. |
|
ABSTRACT
|
|
Introduction: Xeroderma pigmentosum (XP) is an inherited condition characterized by extreme sensitivity to ultraviolet rays from sunlight. It mostly affects the eyes and areas of skin exposed to the sun with some nervous system involvement. Diagnosis is done clinically. The prevalence of that catastrophic disease has been found to be at 1:1,000,00 in the United States and Europe. On the other hand, premature ovarian insufficiency was defined as the development of hypergonadotropic hypogonadism before the age of 40 years in women who have a normal karyotype. Case Report: A 30-year-old female who was a known case of xeroderma pigmentosum since birth. She came to the gynecology clinic complaining of oligomenorrhea for two years. Investigations were done and unfortunately this patient was discovered to have ovarian failure. There is a positive family history of consanguinity. Conclusion: Xeroderma pigmentosum is an autosomal recessive inherited disorder. For that reason, consanguinity is a very important risk factor. There is an increased risk of skin neoplasms and other internal neoplasms such as breast and uterine cancer. Moreover, premature ovarian failure is another rare disorder that we are presenting in this case in association with XP. A study that suggested a relation between XP and ovarian failure had found that three out of twenty XP patients study was found to have POF. In such cases, hormone replacement therapy will be danger as it increases the risk of breast cancer in those patients who are already at risk of internal neoplasms. | |
|
Keywords:
Consanguineous parents, Premature ovarian insufficiency, Xeroderma pigmentosum
| |
|
INTRODUCTION
| ||||||
|
Xeroderma pigmentosum, commonly known as XP, is an inherited condition. It is inherited in an autosomal recessive manner and characterized by an extreme sensitivity to ultraviolet rays from sunlight. This condition mostly affects the eyes and areas of skin exposed to the sun [1]. Some affected individuals also have problems involving the nervous system. The diagnosis of xeroderma pigmentosum is usually done clinically in which three main categories are involved; these are: the skin, the eyes and the nervous system [2][3]. Sixty percent of patients demonstrate acute sun sensitivity in the form of severe sunburn with blistering or persistent erythema on minimal sun exposure. The remaining 40% affected children do not burn easily but developed freckle-like pigmentations. These unusual freckles, when presented on the face before the age of two years, are typical of XP and rarely seen in children with normal DNA repair mechanism [4]. The estimated prevalence of this catastrophic disease has been found to be at 1:1,000,00 in the United States and Europe [5]. A higher prevalence is reported in other populations. In japan, the prevalence has been found to be as high as 1:22,000 [6]. It was found that consanguinity increased the prevalence especially in North Africa (Tunisia, Algeria, Morocco, Libya and Egypt) and the Middle East (Turkey, Israel and Syria) [7][8][9]. On the other hand, premature ovarian insufficiency (POI) is defined as the development of hypergonadotropic hypogonadism before the age of 40 years in women who have a normal karyotype [10]. Premature ovarian insufficiency is considered to be present when a women who is less than 40 years old and had amenorrhea for four months or more, with two serum FSH level in the menopausal range [11][12]. This disorder is sometimes associated with the presence of estrogen deficiency symptoms such as hot flashes, osteoporosis and vaginal dryness. However, in 50–75% of cases ovarian function is intermittent. Accordingly, the absence of vasomotor symptoms, vaginal dryness and other estrogen deficiency symptoms should not make the treating physician exclude the diagnosis of primary ovarian insufficiency [13][14][15]. Premature ovarian insufficiency is a spectrum disease that is mainly characterized by an impairment of ovarian function. This spectrum varies between occult POI and Overt POI. Occult POI is defined as impaired ovarian responsiveness to exogenous or endogenous gonadotropin stimulation despite the presence of regular and predictable ovulatory menstrual cycles. On the other hand, Overt POI is defined as the presence of irregular menses, elevated serum gonadotropins and reduced fertility. We are reporting a 30-year old female who was a known case of xerodrma pigmentosum since childhood. She came to the gynecology clinic complaining of oligomenorrhea for two years. Investigations were done and unfortunately this patient was discovered to have ovarian failure. Literature concerning the relation between these two catastrophic events was reviewed. | ||||||
|
CASE REPORT
| ||||||
|
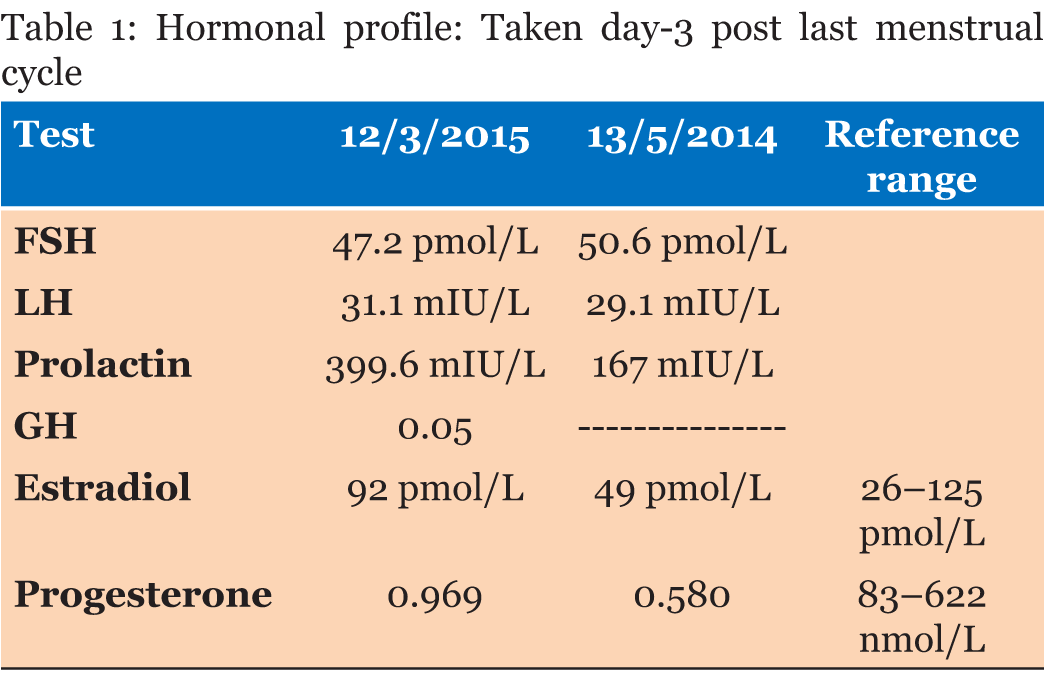
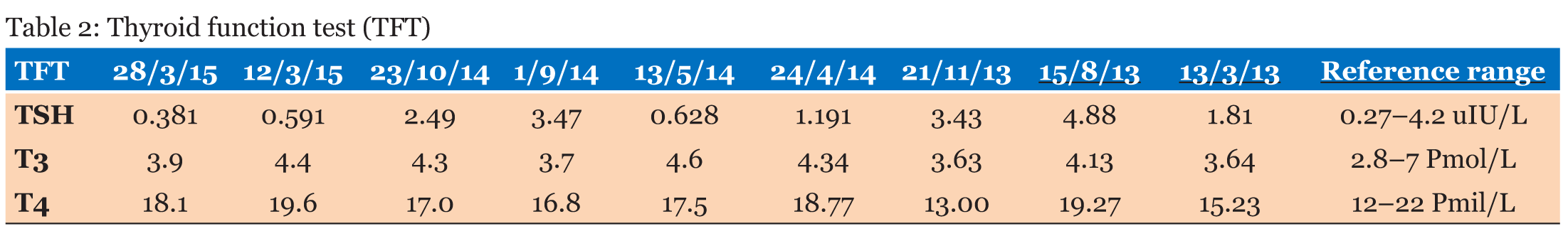
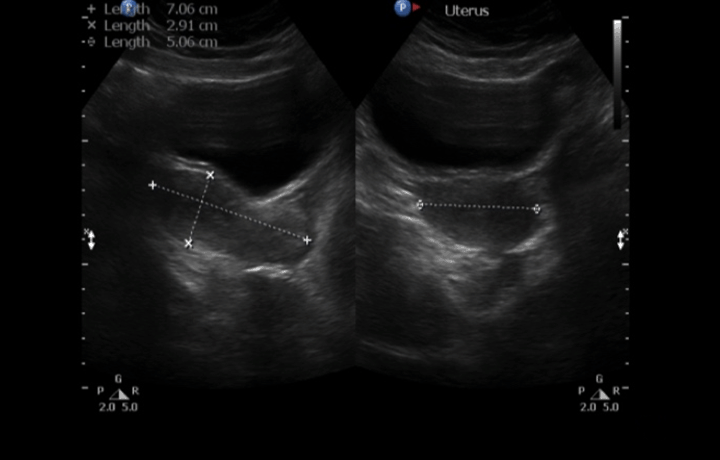
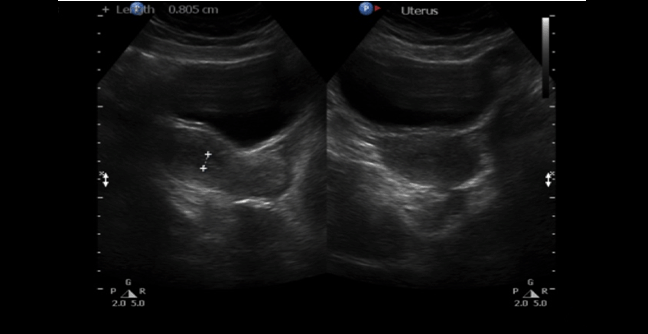
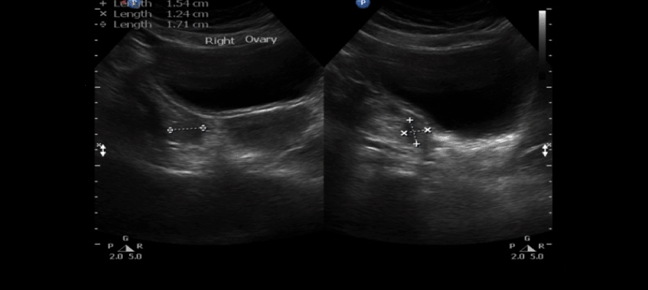
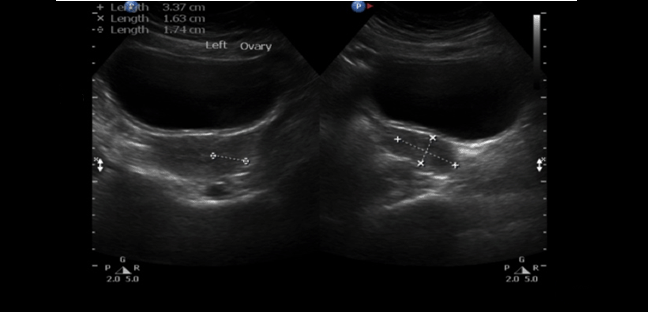
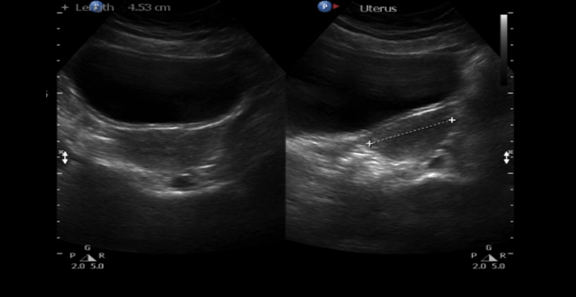
A 30-year-old single female was a known case of xeroderma pigmentosum since childhood. She came to the gynecology clinic complaining of oligomenorrhea. According to her, this problem started two years ago in which she menstruates every three months. She was born with spontaneous vaginal delivery, bottle-fed and showed standard psychosomatic development and menarched when she was 12 years old. Since then, she menstruated regularly with normal amount, color, odor and without any obvious clots. Suddenly, her problem started and the amount of blood became scanty, with no changes in the color, odor or obvious clots formation. She denied dysmenorrhea or metrorrhagia during her regular menstrual periods. Her last menstrual period was two months ago. There was no history of any use of chemotherapy, radiotherapy, steroids or long-term gynecological medications. The patient visited the gynecologist on several occasions and she was prescribed medications that she can not remember for menstrual irregularities. She continued to use them for two months only for they cause her some personality changes as she admitted. She added that she did not menstruate on these medications. Four months ago she started visiting our clinic for the same reason. As a result, investigations were ordered, and Marvelon was prescribed to her once daily. Once again the complaint was not relived and it eventually showed no positive results. Four weeks ago she came again to the hospital for the same issue plus bilateral severe bone aches mainly in the hips and knees that sometimes prevent her from doing any activity. She stated that she had vaginal infection many times and she feels dryness in her vulva. When the patient was diagnosed with xeroderma pigmentosum, her dermatologist advised her to avoid sun exposure and recommended to the use sunscreens. According to her, she was not compliant to that extent. As a complication of xeroderma pigmentosum, she had blindness in both eyes so she had a corneal transplantation bilaterally. Sooner, she became blind in the right eye. Five years ago, her physician found her to have goiter with hypothyroidism. Accordingly, FNA was ordered for her but unfortunately it did not show anything. As a result, FNA was repeated three times but again no results were found. So, the decision was taken to do thyroidectomy. Histopathology showed multinodular goiter. Afterward she was put on thyroxine 100 mg for five days and 50 mg for two days. The patient had radical excision of soft tissue lesions many times as a result of squamous cell carcinoma and malignant melanoma that appeared mainly on the nose. She also had septoplasty as a repair of the nose after the excision. A nasal mass was also excised from the nose. She is continuously on omeprazole and vitamin D. The patient has five siblings (4 males and 1 female). Two of her brothers are affected with the same disease. Her mother menarched at 12 and had normal menstrual cycle since then until she menopaused at 49 years. Her sister also had normal menstrual cycle with no noted irregularities. There is no breast cancer, uterine cancer nor any type of cancers noted among the family. There is a positive history of consanguinity between her parents. She does not smoke and no one of her family does. She studies psychology at the university. When we examined her, Initials including: [height: 151 cm, weight: 77 kg and BMI: 33.7] and vitals including: [blood pressure 120/77, pulse 74 BPM brachial, temperature 37°C oral, respiratory rate 20 and O2 sat 100%]. Generally, the patient looks well, conscious and oriented to person place and time. The freckles are spread on the face, upper limbs, lower limbs, chest, and back. Scars from previous surgeries were noted on the nose, and multiple others were noted on the face. No special facial features are noted, no dysmorphism, no alteration was detected in the fingers and toes. Secondary sexual characteristics are well developed. Neck examination shows no palpable nodes or masses. The abdomen is soft and lax. Chest shows bilateral equal air entry with no added sound and the pericardium has shown normal first and second heart sounds with no obvious added sounds or murmurs. Neurological examination was done and it was unremarkable rather than that she mentioned progressive hearing loss which was not detected on examination. There was neither evident mental impairment nor cognitive deficit. Bone density showed osteopenia. Investigations including hormonal profile, TFT, blood grouping and vitamin D were given in Table 1, Table 2 and Table 3) and Female Pelvic Ultrasound (Figure 1) (Figure 2) (Figure 3) (Figure 4) (Figure 5). The patient is on omeprazole 20 mg orally once daily and vitamin D. She is not following any measures of sun protection. Our plan was to give her hormone replacement therapy but because xeroderma pigmentosum patients have a high risk of internal cancers such as uterine and breast cancers the risk will increase more with the use of HRTs so the role will be by treating the symptoms of premature ovarian inefficiency. On the other hand, she requires a condensed education regarding her issue as she had multiple occasions of skin cancer that were treated with skin excision. | ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
|
DISCUSSION
| ||||||
|
Xeroderma pigmentosum (XP) is known to be an autosomal recessive inherited disorder. One other rare subtype of the disease that is called XP dominant type is inherited as an autosomal dominant disorder [16]. For that reason consanguinity in the parents of the patients of XP has been shown to be an important etiologic factor [17]. According to a study, it has been reported to be 92.8% in XP patients in Libya [18]. Studies from Egypt, Pakistan, and Nigeria have shown a high incidence of the disease as these countries are considered to be among the countries, which has high rates of consanguinity [19][20]. On the contrary, a series of four cases of XP in which consanguinity had not been observed in the parents of those patients have been described [21]. In another case, the patient was born to consanguineous parents. However, there was a varying degree of the disease progression and severity even among sibling who are affected [17]. The patient in the present case also was born to parents who had a history of consanguineous marriage and with variable degrees of affection and progression among the siblings. Her two brothers who are affected with the same disease showed slower progression of the disease with less affection as they never had a history of skin cancer and the pigmentary changes were not involving the same amount of their body as our patient has. Although the disease affect more commonly patients who are the product of consanguineous parents. Stephanic Christen-Zaech et al. reported a case that showed that XP has occurred in an uncle and nephew [22]. In XP, the main defect is in the nucleotide excision repair (NER), in which this leads to deficient repair of DNA damaged by ultraviolet radiation [23]. Nine gene mutations (namely: XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, ERCC1, and POLH) had been reported. As a result, the disease had been classified by complementation groups (XP-A, XP-B, XP-C, XP-D, XP-E, XP-F, XP-F, XP-G). Researches have also described another form of the disease that is called XP variant (XP-V). As with the other subtypes of XP the symptoms and signs of the other subtypes may also be seen in XP-V patients. As a result, the preferred method of laboratory diagnosis is functional testing to screen cells for abnormalities in DNA repair. Simply saying, Xeroderma pigmentosum is mainly diagnosed according to clinical findings and family history [24]. In the presenting case the patient did not do any genetic testing regarding her diagnosis. Diagnosis of XP in our case was conducted according to the clinical findings and the family history in her two brothers. Basically, clinical diagnosis mainly depends on three major areas that are evolved which are the skin, the eyes and the presence of neurological manifestations. According to Bradford in a long-term study of 106 individuals diagnosis depending on skin manifestations can often be made in the first year of life in which about 60% of the affected individuals demonstrated acute sun sensitivity meanwhile the remaining 40% of the affected individuals did not burn easily but instead developed marked skin like pigmentations. This same long-term study has shown that 25% of the affected individuals had characteristic progressive neurologic manifestations that usually worsen slowly and manifest later than skin changes. Of those neurological manifestations, diminished or absent deep tendon reflexes, progressive sensorineural hearing loss, acquired microcephaly and progressive cognitive impairment [2]. Our patient described a history of progressive sensorineural hearing loss. On the other hand, ophthalmologic problems are usually limited to the anterior, ultraviolet-exposed portion of the eye resulting in photophobia, severe keratitis, increased pigmentation in the eyelids with loss of lashes, atrophy of the skin resulting in ectropion and entropion and finally complete loss of the eye lids [3][25]. Unfortunately, our patient had photophobia, severe keratitis, increased pigmentations and finally loss of sight, which was eventually treated by corneal transplantation in both eyes. She started to see again but as a result of her negligence she again lost sight in the left eye. As Bradford mentioned in his long-term study that XP patients has an increased risk of skin cancers namely non-melanoma (basel cell and squamous cell carcinomas) and cutaneous melanoma. The former had a risk that is greater than 10000 folds, which is associated with a median age of onset of nine years in contrast to the other one, which had a risk greater than 200-fold increase and associated with the median age of onset of 22 years [2]. This case had many episodes of radical skin excision due to squamous cell carcinoma and malignant melanoma lesions mainly on the nose, which led to a complete nasal reconstruction. This is not the end as other studies have proven that Xeroderma pigmentosum is also associated with an increased risk of other internal neoplasms mainly gliomas of the brain and spinal cord, tumors of the lung, uterus, breast, pancreas, stomach, kidney, testicles and even leukemia which have been reported in a few individuals with the disease [26][27][28]. Luckily, our patient does not have any of those internal cancers. The patient in this case presented with premature ovarian failure or primary ovarian insufficiency. Basically, no relation has been established regarding those to problems but some studies have looked this up. It has been found that the incidence was approximately 1 in 250 by the age of 35 years and 1 in 100 by the age 40 years. POI’s fully developed form is associated with oligomenorrhea or amenorrhea, symptoms of estrogen deficiency and gonadotropin levels in the menopausal range before the age of 40 [29]. Our patient started facing these menstrual irregularities in an age that is earlier than normal median age for the development of the symptom in other patients as she started to have oligomenorrhea by the age of 27 years. This disorder differs than natural menopause in that menopause is permanent cessation of menstrual periods that is determined after women experience 12 months of amenorrhea without any other obvious pathological or physiological cause and it occurs at a median age of 52.4 years in normal women. Although women with POI experience the symptoms of menopause many patients with spontaneous primary ovarian insufficiency intermittently produce estrogen and ovulate, a few experience intermittent return of regular menses, and in 5–10% of cases, women conceive and have normal pregnancy [30]. Patients with POI present with changes in menstrual function (oligomenorrhea and/or amenorrhea), elevated serum level gonadotropin with low serum estradiol and estrogen deficiency symptoms such as hot flashes and vaginal dryness. As it has been mentioned that intermittent ovarian insufficiency occurs in 50–75% of patients, the absence of vasomotor symptoms or vaginal dryness should not dissuade one from considering the diagnosis in patients with menstrual irregularities [31][32][33]. Some associations have been found to be related to POI of those, prior ovarian surgery chemotherapy or ovarian failure [34]. On the other hand, other autoimmune diseases alone or in combination that may suggest polyglandular autoimmune failure such as hypothyroidism or Graves’ disease, primary adrenal insufficiency, vitiligo, myasthenia gravis, hypoparathyroidism, recurrent mucocutaneous candidiasis or type one diabetes may be associated with autoimmune oophoritis which may occur as part of type 1 or type 2 syndromes of polyglandular autoimmune failure. In a study on 457 women with POI, 15% of those women had chronic lymphocytic thyroiditis [35]. Familial idiopathic forms of ovarian failure have been described in 10% of causes as well as fragile X-syndrome and turner syndrome, which had been, described as common causes of ovarian failure [34][36][37][38]. There is a genetic association in POI as association between the disease and permutation in the FMR1 gene was found. In those cases of familial POI approximately 14% will be found to have mutations in FMR 1 gene. In other sporadic cases, the prevalence of this same permutation is about 2% [39][40][41]. In this case, the patient had a history of an unexplained multinodular goiter and had a total thyroidectomy as a result. Two years later, she started complaining of these menstrual irregularities, which was diagnosed as ovarian failure. Diagnosing POI early is very important as the progression of the disease might result in the development of the known menopause consequences mainly osteoporosis. Although early diagnosis is important, many women experience delayed diagnosis up to five years in 25% of cases [42]. Osteoporosis as a consequence of ovarian failure was evident in our patient as she came to the clinic on the last visit complaining of severe bone aches that were described lately as osteopenia on bone density profile. The relation between premature ovarian failure and xeroderma pigmentosum was not established yet. A study was done from 2004 to 2014 on 20 female patients who are known cases of XP, which aimed at finding the obstetrics and gynecology health in XP patients found that one of those 20 patients had precocious puberty and one had infertility. What was relevant is that 3 out of 20 XP patients had ovarian failure, which means that 15% of them had ovarian failure. This pilot study suggests that there is an increase in the risk of premature ovarian insufficiency, precocious puberty, possible increased risk of neoplasm growth during pregnancy and an increased risk of pregnancy complications. According to this study, a patient was found to have vulvar dysplastic nevus. This finding emphasizes the need of including the external genitalia as part of the routine skin examination in those patients. | ||||||
|
CONCLUSION
| ||||||
|
As xeroderma pigmentosum is considered a genetically inherited disorder, premature ovarian insufficiency is sometimes caused by mutations in some genes. This emphasizes the need for genetic counseling and excessive genetic studies in both diseases until a relation between them are established. | ||||||
|
REFERENCES
| ||||||
| ||||||


|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Abdullah K. Agabawi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Wafa M. Faqeeh – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Amal A. Kokandi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Hisham Nassief – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of Submission
The corresponding author is the guarantor of submission. |
|
Source of Support
None |
|
Conflict of Interest
Authors declare no conflict of interest. |
|
Copyright
© 2018 Abdullah K. Agabawi et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|