 |
Review Article
Prenatal diagnosis, management, and outcomes of urinary tract anomalies
1 Department of Obstetrics and Gynecology, Division of Maternal Fetal Medicine, University of Mississippi Medical Center, Jackson, Mississippi 39216, USA
Address correspondence to:
Sarah Araji
2500 North State Street, Jackson, MS 39216,
USA
Message to Corresponding Author
Article ID: 100169Z08KM2024
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Missling KS, Sullivan KS, Araji S. Prenatal diagnosis, management, and outcomes of urinary tract anomalies. J Case Rep Images Obstet Gynecol 2024;10(1):11–22.ABSTRACT
Congenital urinary tract anomalies are one of the more common defects noted on prenatal ultrasound. There are a variety of anomalies that can occur with a broad spectrum of outcomes with different degrees of severity based on pathologic processes. Ultrasound is the imaging modality utilized to allow visualization of the urinary tract system to diagnose these anomalies. We provide a review of these classifications with imaging and diagnostic recommendations, as well as epidemiology and associated defects. The aim of this review is to bring a clinically relevant and succinct understanding of congenital urinary tract anomalies and considerations for their various pathologies.
Keywords: Congenital, Fetal, Ultrasound, Urinary tract
Introduction
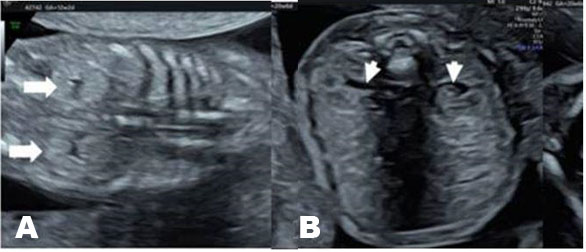
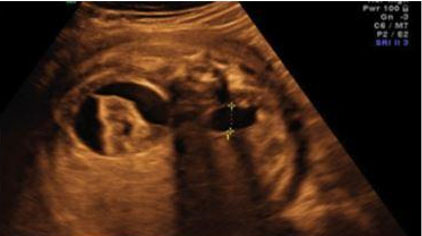
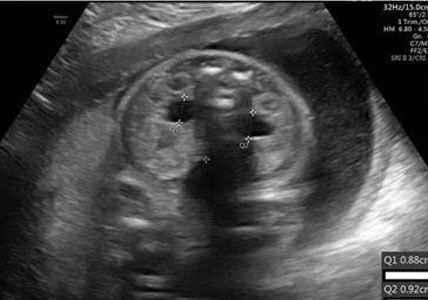
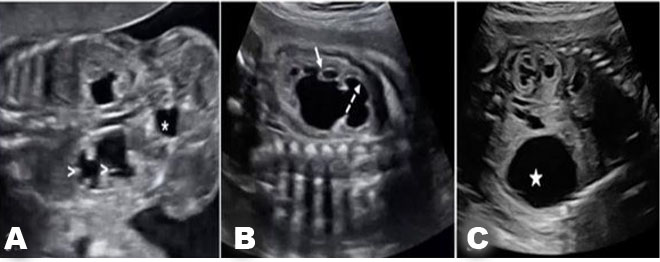
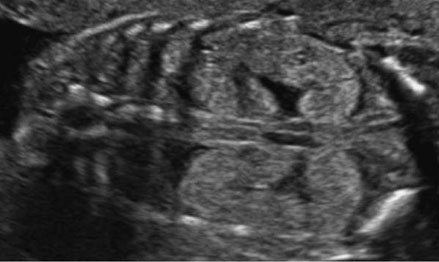
The fetal urinary tract has a very intricate development process and is the most common system affected by congenital defects [1]. Congenital anomalies of the kidneys and the urinary tract (CAKUT) account for approximately 30% of all congenital malformations. Abnormalities can affect the upper urinary tract such as the kidneys and the ureters and the lower urinary tract, impacting the bladder and the urethra which will then affect the amniotic fluid volume. Congenital anomalies of the kidneys and the urinary tract causes include genetic etiologies, maternal diet, and fetal exposures [2]. The antenatal detection of the anomalies, which is at 73.87%, has helped improve outcomes [3]. The most used imaging tool is ultrasound, furthermore it is standard practice to visualize the kidneys and the bladder during the second trimester ultrasound in pregnancy (Figure 1A and Figure 1B) [4]. The spectrum of outcomes for these patients ranges from no clinical significance to more serious outcomes such as pulmonary hypoplasia and end-stage renal disease [1],[5]. While each malformation has a different male to female ratio, CAKUT impacts more males [3]. A multidisciplinary care team is important when diagnosing, managing, and treating CAKUT. In this review, we will discuss the relevant embryological process and discuss different anomalies (Figure 1A and Figure 1B).
Embryology
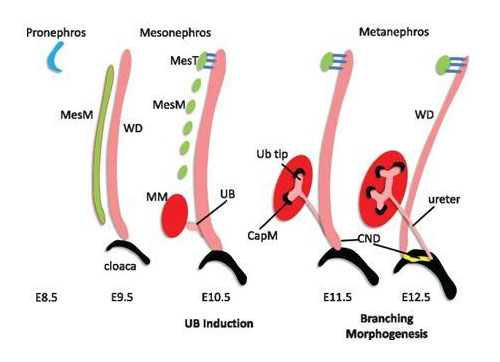
Kidney development is a multistage process that begins in the anterior tract and then moves posteriorly [1]. First, the nephric duct forms (embryonic day 22 in humans) in the intermediate mesoderm inducing kidney development in three stages: the pronephros, the mesonephros, and the metanephros [1],[6] (Figure 2). The pronephros and mesonephros degenerate while the metanephros gives rise the mature kidneys via a series of divisions and branching, which can be detected at the fifth or sixth week of gestation. The ureteric bud arises as an outpouching from the nephric duct which becomes the ureter. By the eighth week of gestation the fetal kidneys will have ascended from the pelvis to the retroperitoneal fossa and rotated medially 90° [6].
The fetal kidneys make and release urine via a patent urethra by eight weeks gestation with fetal swallowing beginning shortly thereafter; however, it is not until about 16 weeks gestation that fetal urine is the primary source of amniotic fluid [7]. By the second half of pregnancy, fetal urinary production and fetal swallowing are key contributors in the total amniotic fluid index. The major kidney function antenatally is the preservation of a normal amniotic fluid volume via urinary production. Prenatally kidneys do not contribute to acid-base or electrolyte balance; however, the amniotic fluid volume is an indicator of kidney function (Figure 2).
Below we will discuss the following urinary tract abnormalities:
- Renal agenesis
- Horseshoe kidney
- Urinoma
- Pyelectasis
- Duplicated (duplex) collecting system
- Hydroureter
- Ureteropelvic junction (UPJ) obstruction
- Lower urinary tract obstruction (LUTO)
- Multicystic dysplastic kidney (MCDK)
- Autosomal recessive polycystic kidney disease (ARPCKD)
Renal agenesis
Terminology
Ureteric bud fails to fuse leading to the congenital absence of one or both kidneys [9]. An important distinction must be made between unilateral and bilateral in terms of treatment and outcome.
Imaging
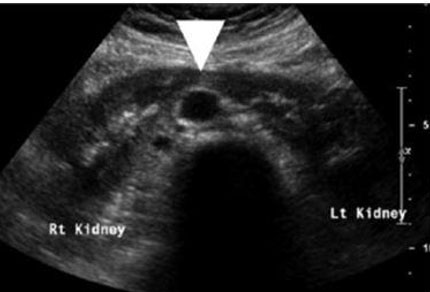
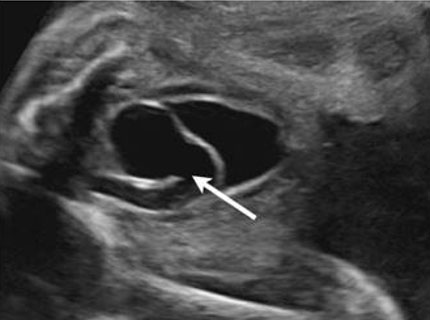
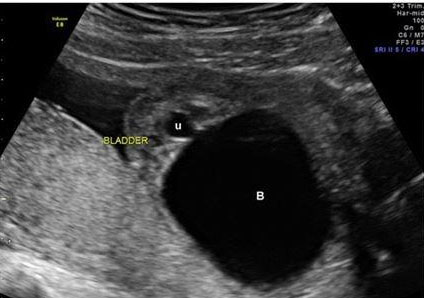
Kidneys can be visualized on ultrasound as early as 12 weeks gestation. However, unilateral or bilateral renal agenesis is most often discovered during an anatomy ultrasound examination at 18–22 weeks gestation. Key imaging findings include an empty renal fossa with the absence of kidneys in axial, sagittal, and coronal planes at the spine level and below the stomach. The “lying-down” adrenal sign, which is a large and flat appearance of the adrenal glands, an absence of the renal artery at a 90° angle in the coronal plane, and an absence of the fetal bladder are all key diagnostic features that may be visualized (Figure 3). Assessment for a pelvic or ectopic kidney should be performed if unilateral renal agenesis is suspected. Bilateral renal agenesis (RA) is suspected when the bladder cannot be visualized in the setting of anhydramnios.
An abnormal amniotic fluid index is not seen in the first trimester due to amniotic volume being placental in origin until 16–19 weeks of gestation. The lack of amniotic fluid may make visualization through ultrasound difficult. In these cases, diagnostic amnioinfusion can be used as a technique to visually confirm the diagnosis.
Differential diagnosis
- Ectopic or pelvic kidneys
- Preterm premature rupture of membranes
- Placental insufficiency
- Multicystic dysplastic kidney
- Autosomal recessive polycystic kidney disease
Clinical issues and prognosis
Unilateral renal agenesis is usually incidentally found during routine ultrasound examination and may not be discovered until adulthood in some cases. The prevalence of RA is estimated to be 0.03% with unilateral RA representing 48% of all cases and bilateral renal agenesis comprising 52% of cases [10]. It should be noted that bilateral RA occurs three times more often in males than females [11].
In unilateral RA, the remaining kidney is often larger and more susceptible to injury, ischemia, and toxicity. In 50% of cases, patients develop hypertension and may be more likely to develop proteinuria and renal insufficiency [11].
Bilateral RA is a lethal diagnosis due to failure of pulmonary development in the absence of amniotic fluid. Thirty-three percent of pregnancies end in stillborn while survivors die of respiratory failure as a result of pulmonary hypoplasia [11].
Prenatal diagnostic testing should be offered. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Diagnostic checklist
Describe in detail whether agenesis is bilateral or unilateral. Describe if and where there is hypoplasia or agenesis. Note location and any possible defects of the urethra.
Describe any other abnormalities and look for possible associated and related anomalies [12].
If diagnosis of unilateral RA was made in the 2nd trimester, perform a 3rd trimester ultrasound follow-up. If bilateral RA is suspected, consider a magnetic resonance imaging (MRI) to get a specific diagnosis, identifying the underlying cause of oligohydramnios to appropriately counsel parents along with palliative care and pediatric urology (Figure 3).
Horseshoe kidney
Terminology
Abnormal fusion and ascent of the kidneys causing a horseshoe appearance.
Imaging
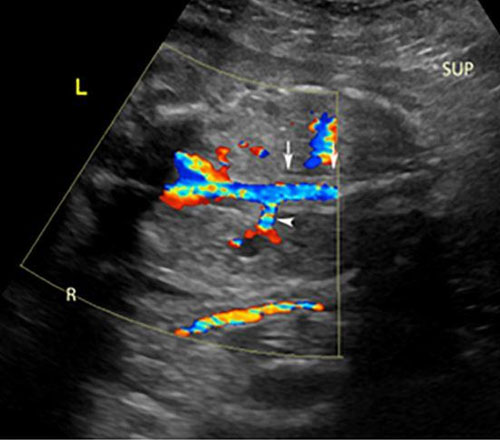
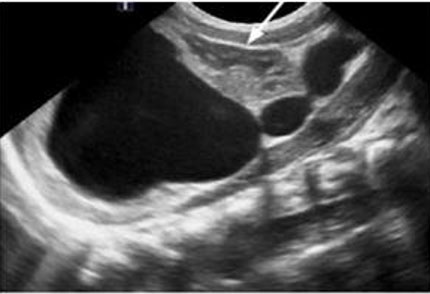
Horseshoe kidneys are often identified incidentally by a variety of imaging modalities such as ultrasound, CT, or MRI. Third trimester evaluation of the fetal kidneys in the coronal plane will provide the most accurate visualization of kidney orientation. Ultrasound evaluation may reveal fusion of medially rotated lower poles connected by bridging tissue called the isthmus (Figure 4). Horseshoe kidneys are usually found lower within the pelvis than the normal anatomic location and are typically bent or curved. Color Doppler will indicate a variance in blood supply. Magnetic resonance imaging is rarely needed unless ultrasound is inconclusive.
Differential diagnosis
- Crossed fused renal ectopia
Clinical issues and prognosis
While horseshoe kidney may be the etiology of unilateral renal fossa or ureteropelvic junction obstruction, it is most often discovered incidentally. The incidence of horseshoe kidney in the general population is 1 in 500 [13]. There is a noted male predominance of approximately 2:1 [13]. Horseshoe kidney is most often clinically silent. The most common complication is urolithiasis and frequent urinary tract infections. About one-third of patients have other anomalies due to a syndrome [14]. These patients will have varying prognosis dependent on the severity of the associated syndrome. Horseshoe kidney is commonly seen in Turner syndrome. Prenatal diagnostic testing is recommended. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
After prenatal diagnosis, postnatal renal ultrasound should be performed to confirm the diagnosis; consider visualization of uterus in females.
Diagnostic checklist
Medial orientation of the lower pole may be the best indicator of horseshoe kidney if the isthmus is thin. Consider and look for associated anomalies (Figure 4).
Urinoma
Terminology
An encapsulated collection of urine around the kidney resulting from leakage of urine secondary to bladder rupture or injury to renal parenchyma.
Imaging
An anechoic fluid filled structure is visualized adjacent to the fetal kidney (Figure 5). This fluid collection can put pressure on the kidneys, displacing them from the usual anatomic location. Identifying normal anatomical structures to delineate the location of the fluid collection can help differentiate a urinoma from other pathologies with similar sonographic appearance.
Differential diagnosis
- Hemorrhagic neuroblastoma
- Lymphangioma
- Gastrointestinal system-related pathologies forming cysts
- Medullary cystic kidney disease (MCKD)
- Uteropelvic junction (UPJ) obstruction
Clinical issues and prognosis
Urinomas are often the result of a urinary tract pathology causing severe obstruction, most commonly posterior urethral valves (PUV) and UPJ obstruction [15],[16]. The workup and prognosis for fetal urinomas are determined by the primary anomaly. Anhydramnios is associated with a poor prognosis. Moreover, irreversible ipsilateral renal damage is seen in 75% of cases [17]. Both resolution of a urinoma and lack of growth are signs of fetal kidney dysfunction. Prenatal diagnostic is reasonable and is determined by the underlying anomaly. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
Detailed ultrasound, identification of the cause of the urinoma, pediatric urology consultation, and close follow-up for the ultrasound evolution of the findings are recommended. Magnetic resonance imaging may be helpful in better evaluating the renal parenchyma [15]. Drainage of a urinoma has only been helpful to avoid abdominal dystocia with vaginal delivery; however, it does not improve outcomes [15],[17].
Diagnostic checklist
Look for sonographic signs of obstructive uropathy to identify the source of the urinoma.
Serial ultrasound to evaluate the amniotic fluid index to determine contralateral kidney function (Figure 5).
Pyelectasis
Terminology
Distension of the renal pelvis without calyceal distension. Pyelectasis is most often a transient, isolated finding. Hydronephrosis has a similar definition to pyelectasis but is differentiated by larger measurements of the fluid-filled space [18].
Imaging
Ultrasound findings for pyelectasis are most often bilateral [19] (Figure 6). Pyelectasis is diagnosed by measuring the antero-posterior (AP) diameter of the renal in an axial plane. Longitudinal views are helpful to rule out hydronephrosis and identify a distended ureter if present.
Urinary tract dilation (UTD) A1: AP diameter 4<7 mm (<27 weeks), 7–<10 mm (>27 weeks).
UTDA2: ≥7 m (<27 weeks), ≥10 mm (≥27 weeks) or any calyceal or ureteral dilation.
Differential diagnosis
- Ureteropelvic junction obstruction
- Bladder outlet obstruction
- Ureterovesical junction obstruction
Clinical issues
Pyelectasis is found incidentally through ultrasound in low-risk patients and is most often found in normal fetuses. However, pyelectasis can also be found with aneuploidies and is considered a soft marker for trisomy 21 [20]. Pyelectasis is more likely to progress to hydronephrosis if it is unilateral. Aneuploidy screening is recommended, prenatal diagnostic testing is reasonable especially if other anomalies are seen.
Treatment
Mild pyelectasis does not require any treatment, just monitoring with serial ultrasonography to evaluate for progression. Obstructive lesions will most likely need some form of surgical intervention.
Diagnostic checklist
Repeat ultrasound imaging in 4–8 weeks to look for progression. Postnatal ultrasound may also be helpful in establishing prognosis. Because of its association with pyelectasis, look for other soft markers of aneuploidy. To rule out reflux, consider a voiding cystourethrogram postnatally [18] (Figure 6).
Duplicated (duplex) collecting system
Terminology
A kidney with two renal pelves as a result of duplication of the ureteric bud. There are two subtypes: complete duplication in which each renal pelvis has a ureter that attaches to the bladder and partial duplication, the more common subtype, in which the two ureters fuse before inserting into the bladder.
Imaging
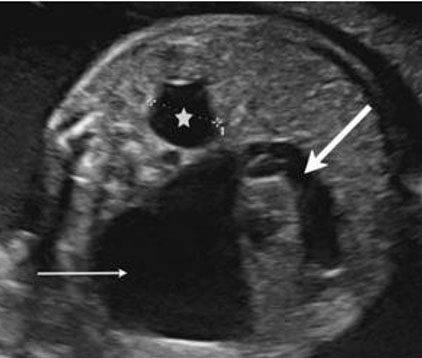
The best diagnostic clue is a dilated upper collecting system and a ureterocele within the urinary bladder (Figure 7). Kidneys should be evaluated in transverse and longitudinal planes (Figure 8).
A duplex kidney has two renal pelves and is larger than normal. It can be bilateral in up to 30% of cases [9].
The ureterocele is seen as a cystic structure within the fetal urinary bladder. The ureterocele is associated with the ectopic insertion of the ureter draining the upper pelvis.
Differential diagnosis
- Pyelectasis
- Ureteropelvic junction (UPJ) obstruction
- Vesicoureteral reflux
- Isolated ureterocele
- Megaureter
Clinical issues and prognosis
Duplex kidney is the most common congenital anomaly of the urinary tract with a prevalence of 0.8% at autopsy [21],[22]. The ureter from the lower pelvis enters the bladder in the trigone and the ureter from the upper pelvis can be inserted at the trigone or be ectopic. Depending on the location of the ectopic insertion, this can lead to obstruction or vesicoureteral reflux. The upper pelvis will become dilated secondary to obstruction whereas the lower pelvis is at risk for vesicoureteral reflux. Duplex kidney is sporadic and is rarely associated with aneuploidies or a genetic anomaly in the absence of other anomalies unless a strong family history is present.
Treatment
Routine obstetric follow-up is recommended with ultrasound follow-up to evaluate for obstruction and amniotic fluid. In utero treatment is usually not indicated. Pediatric urology consult is recommended for counseling antenatally.
Management after delivery includes urology consult, renal and bladder ultrasound, voiding cystourethrogram (VCUG) to evaluate the ureterocele. Surgery is indicated depending on the severity of the abnormality. Incision of the ureterocele is indicated in the setting of obstruction or infection.
Diagnostic checklist
Weigert–Meyer rule: Ectopic upper pole ureter insertion inferior and medial to normotopic ureter in trigone of the bladder, usually associated with ureterocele in bladder. Upper pelvis dilated from obstruction, lower pelvis refluxes (Figure 7 and Figure 8).
Hydroureter
Terminology
Hydroureter refers to the dilation of the ureter due to obstruction of urine outflow. Obstruction can be caused by urethral stricture and stenosis of the ureterovesical or uteropelvic junction [23]. Megaureter refers to a ureter that is =7 mm in diameter after 30 weeks of gestation [24].
Imaging
The hydroureter will present as tortuous and tubular in the fetal pelvis, the presence of a dilated ureter on ultrasound is an abnormal finding (Figure 9). The hydroureter may communicate with the kidney, bladder, or both.
Differential diagnosis
- Ureteropelvic junction (UPJ) obstruction
- Ureterovesical junction (UVJ) obstruction
- Bladder outlet obstruction
Clinical issues and prognosis
Hydroureter can be an isolated anomaly but is usually found with other genitourinary anomalies. It is most often associated with vesicoureteral reflux and lower urinary tract obstruction, such as bladder outlet obstruction or duplicated collecting system. Prognosis for hydroureter is dependent on the severity of obstruction, associated anomalies and amniotic fluid volume. An isolated megaureter can be managed without surgical intervention in three-quarters of infants [25]. However, infants with a severe obstruction may require dialysis after birth because of renal failure [25]. Prenatal diagnostic testing should be offered especially in the setting of multiple anomalies. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Diagnostic checklist
In order to differentiate between a dilated loop of bowel and hydroureter, observe bowel peristalsis [25]. If a unilateral hydroureter is found, examine the bladder and contralateral collecting system. Follow up with serial ultrasound examinations. It may also be beneficial to consult pediatric nephrology or urology to form a plan on postnatal evaluation (Figure 9).
Ureteropelvic junction obstruction (UPJO)
Terminology
Upper urinary tract obstruction at the UPJ leading to congenital hydronephrosis.
Imaging
Ultrasound findings include renal pelvis dilation that ends at UPJ. Calyceal dilation may be also noted (Figure 10). Kidney enlargement is also a hallmark of UPJO. If the obstruction is severe, dysplasia with increased echogenicity [26]. Normal ureters are usually found if UPJ obstruction is unilateral. Ureteropelvic junction obstruction can be associated with contralateral abnormalities, bilateral obstructions, and extrarenal anomalies [26].
Differential diagnosis
- Multicystic dysplastic kidney
- Mild pyelectasis
- Megaureter
- Vesicoureteral reflux
Clinical issues and prognosis
UPJ obstruction is usually incidentally found in ultrasound. It is more frequently found in males than females [26]. The left side is twice as likely to be affected as the right side [27]. Hydronephrosis is caused by UPJO 80% of the time [27]. Ureteropelvic junction obstruction has an incidence of an estimated 1 in 1000 to 1500 births [27]. Unilateral UPJ obstruction has an excellent prognosis as many resolve spontaneously and do not need treatment. Corrective surgery may be indicated if renal function is impaired. Prenatal diagnostic testing is recommended. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
Rarely will there is prenatal intervention because mild UPJO may resolve spontaneously without treatment. If renal function is impaired, corrective surgery may be considered such as pyeloplasty, endopyelotomy, and percutaneous drainage [27].
Diagnostic checklist
Recommend follow-up ultrasounds at 4–6 week intervals: UPJO may progress rapidly, monitor contralateral kidney. Postnatal renal ultrasound is recommended 72 hours after delivery. Severe UPJO may look like renal cysts, so make sure that the cysts noted connect with renal pelvis (Figure 10).
Lower urinary tract obstruction (LUTO)
Terminology
Obstruction in the lower urinary tract preventing urinary excretion. There are multiple anomalies that fall under LUTO.
Imaging
Diagnosis of LUTO is accurately done through ultrasound with high sensitivity and specificity [28]. Lower urinary tract obstruction may present in fetuses with a dilated renal pelvis and small fluid filled areas (Figure 11). Distinctions between obstructive and non-obstructive causes can be made through echogenicity and presence of oligohydramnios [29]. Obstructions may present with a large bladder (megacystis) and dilation of urethra (“keyhole sign”) depending on location of obstruction [30]. If anhydramnios is noted on ultrasound it is associated with a higher prevalence of pulmonary hypoplasia and renal dysplasia [28]. Changes in renal parenchyma on ultrasound are also associated with a higher rate of renal dysplasia [28]. Magnetic resonance imaging is suggested if ultrasound cannot be used for assessment of genitourinary tract because of oligohydramnios. Fetal serial urine analysis will be helpful in understanding renal function and provide further information for prognosis.
Clinical issues and prognosis
Lower urinary tract obstruction affects an estimated 2.2 per 10,000 births [31]. The pathological process that contributes to LUTO is mostly unknown. The two most common presentations of LUTO are posterior urethral valve (PUV) atresia affecting about 64% and urethral atresia (39%) [28]. Males are more commonly affected than females; however, females with LUTO will present with more complex and lethal pathologies [28]. Lower urinary tract obstruction is associated with high mortality and morbidity. With oligohydramnios, mortality was measured to be as high as 80% and 25–30% surviving fetuses develop end-stage renal failure [28]. Prenatal diagnostic testing should be offered, fluid can be obtained by vesicocentesis in cases of anhydramnios or oligohydramnios. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
Most surgical intervention is utilized immediately during the postnatal period [32]. There are several techniques that can be used in utero to relieve the obstruction such as percutaneous vesico-amniotic shunting or cytoscopically placed urethral vesico-amniotic shunts and ablations of posterior urethral valves. This has been shown to improve survival in some patients, however, morbidity associated with renal failure continues to be an issue later in life [33].
Diagnostic checklist
It is important in diagnosis to visualize the bladder and kidneys assessing for any abnormalities. Identifying specific obstruction will be key in prognosis and consideration for surgical intervention (Figure 11).
Multicystic dysplastic kidney (MCDK)
Terminology
The replacement of renal parenchyma with cysts is also known as renal cystic dysplasia.
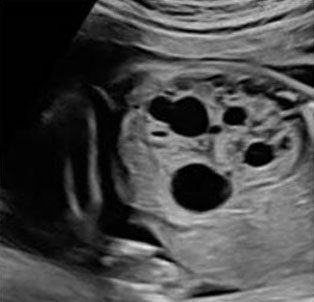
Imaging
The best diagnostic clue for MCDK is multiple, different sized cysts seen in the renal fossa. Multicystic dysplastic kidney presentation can vary greatly from the size of the kidney, laterality, and associated anomalies (Figure 12). More often non-communicating cysts are found only in the left kidney with an increased renal size and loss of reniform shape [34]. Bilateral MCDK is associated with poor prognosis. In 40% of cases a non-MCDK anomaly is found in the contralateral kidney [11].
Axial and longitudinal views of the kidney as a part of routine anatomy scans are best for identifying MCDK. Follow-up every 3–4 weeks is recommended to watch contralateral kidney for development of anomalies and monitor the size of the cysts as they may increase. Amniotic fluid should also be followed very carefully as that could give indicators to contralateral anomalies. Magnetic resonance imaging may be helpful in complicated cases.
Differential diagnosis
- Ureteropelvic junction obstruction
- Obstructive cystic dysplasia
- Autosomal recessive polycystic kidney disease
- Autosomal dominant polycystic kidney disease
Clinical issues and prognosis
Multicystic dysplastic kidney can be diagnosed during an antenatal anatomy scan incidentally. The incidence of unilateral MCDK is 1:1000 [11]. The incidence of bilateral MCDK is 1:10,000 [34]. The male to female ratio is 2:1 [9].
Unilateral MCDK has an overall good prognosis dependent on function of contralateral kidney. The affected kidney in 90% of cases is nonfunctioning and is increasingly likely to involute with time [11]. There are some rare complications such as infection and hypertension. The contralateral kidney may have compensatory hypertrophy. Bilateral MCDK is almost always fatal. Severe contralateral anomalies can be fatal or lead to renal insufficiency, while mild anomalies can be correctable with a good prognosis [9]. Amniocentesis is recommended when MCKD is diagnosed, chromosomal anomalies are more frequent if other anomalies are seen. Syndromes such as Meckel–Gruber, Trisomy 13 and 18 can also be associated with MCDK and can guide the molecular testing depending on the ultrasound findings. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
It is recommended to determine if MCKD is isolated. If the condition isn’t isolated, it’s advisable to undergo amniocentesis followed by comprehensive genetic testing and counseling to ascertain any potential associated syndromes.
For MCDK, the neonatal workup requires an ultrasound for diagnosis and voiding urethrocystogram to evaluate reflux. Multicystic dysplastic kidney can present neonatally as a palpable mass and patients can show symptoms of contralateral ureteropelvic junction obstruction.
An isotope renal scan can assess function. It is recommended to perform an ultrasound every 6 months for 1 year after diagnosis and yearly until kidney involution. Complications that could arise such as infections, hypertension, and Wilms tumor may indicate surgical excisions. Pregnancy termination can be offered for bilateral MCDK.
Diagnostic checklist
In MCDK, it is important to evaluate the function of the contralateral kidney with follow-up ultrasounds and amniotic fluid measurements. It is also important to determine that cysts do not communicate. There may be a presentation of “hydronephrotic type” of MCDK that shows large cysts surrounded by smaller cysts [9]. If a cystic mass in the pelvis is visualized, consider MCDK in a pelvic kidney (Figure 12).
Autosomal recessive polycystic kidney disease (ARPCKD)
Terminology
Autosomal recessive polycystic kidney disease (ARPCKD) is a single gene disorder resulting in bilateral, symmetric, cystic renal disease. It affects the medulla specifically the distal convoluted tubules and collecting ducts. Autosomal dominant polycystic kidney disease (ADPCKD) is asymmetric renal enlargement and cysts are sometimes seen in late 3rd trimester [4]. Autosomal recessive polycystic kidney disease has a different presentation than ARPCKD with normal amniotic fluid and kidney echogenicity; however, it rarely presents in fetuses so this section will focus on ARPCKD.
Imaging
On ultrasound, kidney size will be large for gestational age. Kidneys can become 3–10 times the normal size but may not enlarge until mid-second trimester [35] . A key diagnostic feature is diffusely echogenic kidneys. Notably, ultrasound findings will show a normal hypoechoic cortex and thin hypoechoic rim around echogenic medulla (Figure 13) [11]. This feature may be difficult to visualize in severe disease. Autosomal recessive polycystic kidney disease may also present with oligohydramnios, where the fetal bladder may not be visible. Indication of oligohydramnios is concerning for grim prognosis. Pulmonary hypoplasia, in which the chest is smaller in relation to the abdomen, may also be seen and be indicative of poor prognosis. On ultrasound, small discrete cysts may be visible, but do not predominate [11]. An MRI study will help with ambiguous ultrasound findings and will present similar, notable features.
Differential diagnosis
- Trisomy 13
- Meckel–Gruber syndrome
- Beckwith–Wiedemann
- Bilateral multicystic dysplastic kidney
- Bardet–Biedl syndrome
Clinical issues and prognosis
The majority of ARPCKD cases are detected before 24 weeks gestation. The incidence of ARPCKD is 1 in 6,000 to 55,000 [36].
The phenotype of polycystic kidney disease is dependent on genetic factors. Autosomal recessive polycystic kidney disease is due to variants in PKHD1 gene. Disease severity depends on gene expression in families, but also will vary within a family. Autosomal recessive polycystic kidney disease comes in three types of forms: perinatal, neonatal, infantile, and juvenile. If a fetus presents early with ARPCKD then the most likely outcome due to oligohydramnios and pulmonary hypoplasia is stillbirth or neonatal death [37]. The perinatal form also has a high death rate and fetuses often suffer from severe renal disease, pulmonary hypoplasia, and minimal hepatic fibrosis [37]. The juvenile form is milder, affecting 1 in 5,000 persons and will present with minimal renal disease and marked hepatic fibrosis [36]. Liver disease and hypertension are common in survivors [35]. Renal and overall survival decreases over age. The recurrence risk in future pregnancies for ARPCKD is 25% [37].
Prenatal diagnostic testing should be offered, chromosomal microarray analysis is helpful when there are additional ultrasound findings; however, the goal is to identify variants in the PKHD1 gene. Further testing such as whole exome sequencing should be considered based on the whole clinical picture.
Treatment
Genetic counseling is key if renal disease is prevalent in the family [35] with options for assisted reproductive technology and pre-implantation diagnostic testing discussed. Termination of pregnancy is offered because of high mortality and morbidity rates in affected fetuses [35]. If pregnancy is continued, consider palliative care consult.
Diagnostic checklist
If parents are known carriers, it is recommended to obtain several measurements of kidneys in the fetus. It is recommended to monitor amniotic fluid for oligohydramnios and measure thoracic circumference for pulmonary hypoplasia (Figure 13).
Conclusion
Congenital urinary tract anomalies encompass a broad range of abnormalities affecting any part or parts of the urinary tract system. Anomalies can vary in presentation, pathology, and prognosis. A detailed prenatal ultrasound is most often the mode of discovery of these abnormalities and is vital to the visualization of affected structures. Information gained through ultrasound and genetic testing is used in the management of cases with multidisciplinary teams and can improve outcomes. This review classifies anomalies, describes typical ultrasound findings, epidemiology, and diagnostic considerations to give a clinically relevant overview of urinary tract anomalies.
REFERENCES
1.
Stonebrook E, Hoff M, Spencer JD. Congenital anomalies of the kidney and urinary tract: A clinical review. Curr Treat Options Pediatr 2019;5(3):223–35. [CrossRef]
[Pubmed]

2.
Murugapoopathy V, Gupta IR. A primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol 2020;15(5):723–31. [CrossRef]
[Pubmed]
3.
Li ZY, Chen YM, Qiu LQ, et al. Prevalence, types, and malformations in congenital anomalies of the kidney and urinary tract in newborns: A retrospective hospital-based study. Ital J Pediatr 2019;45(1):50. [CrossRef]
[Pubmed]
4.
Janjua HS, Lam SK, Gupta V, Krishna S. Congenital anomalies of the kidneys, collecting system, bladder, and urethra. Pediatr Rev 2019;40(12):619–26. [CrossRef]
[Pubmed]
5.
Mileto A, Itani M, Katz DS, et al. Fetal urinary tract anomalies: Review of pathophysiology, imaging, and management. AJR Am J Roentgenol 2018;210(5):1010–21. [CrossRef]
[Pubmed]
6.
Talati AN, Webster CM, Vora NL. Prenatal genetic considerations of Congenital Anomalies of The Kidney and Urinary Tract (CAKUT). Prenat Diagn 2019;39(9):679–92. [CrossRef]
[Pubmed]
7.
Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: Not just fetal urine anymore. J Perinatol 2005;25(5):341–8. [CrossRef]
[Pubmed]
8.
Jain S, Chen F. Developmental pathology of congenital kidney and urinary tract anomalies. Clin Kidney J 2018;12(3):382–99. [CrossRef]
[Pubmed]
9.
Society for Maternal-Fetal Medicine (SMFM). Electronic address: pubs@smfm.org; Norton ME, Cheng Y, et al. SMFM Fetal Anomalies Consult Series #4: Genitourinary anomalies. Am J Obstet Gynecol 2021;225(5):B2–35. [CrossRef]
[Pubmed]
10.
Plutecki D, Koziol T, Bonczar M, et al. Renal agenesis: A meta-analysis of its prevalence and clinical characteristics based on 15641184 patients. Nephrology (Carlton) 2023;28(10):525–33. [CrossRef]
[Pubmed]
11.
Woodward PJ, Kennedy A, Sohaey R. Diagnostic Imaging. 4ed. Philadelphia: Elsevier; 2021.
12.
Division of Birth Defects and Developmental Disabilities, NCBDDD, Centers for Disease Control and Prevention. [Available at: https://www.cdc.gov/ncbddd/birthdefects/surveillancemanual/quick-reference-handbook/renal-agenesis-hypoplasia.html]
13.
Kirkpatrick JJ, Leslie SW. Horseshoe Kidney. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
[Pubmed]
14.
Boatman DL, Kölln CP, Flocks RH. Congenital anomalies associated with horseshoe kidney. J Urol 1972;107(2):205–7. [CrossRef]
[Pubmed]
15.
Yitta S, Saadai P, Filly RA. The fetal urinoma revisited. J Ultrasound Med 2014;33(1):161–6. [CrossRef]
[Pubmed]
16.
Lundar L, Aksnes G, Mørkrid L, Emblem R. Prenatal extravasation of urine seems to preserve renal function in boys with posterior urethral valves. J Pediatr Urol 2019;15(3):241.e1–7. [CrossRef]
[Pubmed]
17.
Balcom AH, Pircon R, Worthington D, Carr M. Spontaneous resolution of an in utero perirenal urinoma associated with posterior urethral valves. Urology 1999;54(2):366–7. [CrossRef]
[Pubmed]
18.
Persutte WH, Koyle M, Lenke RR, Klas J, Ryan C, Hobbins JC. Mild pyelectasis ascertained with prenatal ultrasonography is pediatrically significant. Ultrasound Obstet Gynecol 1997;10(1):12–8. [CrossRef]
[Pubmed]
19.
Hindryckx A, De Catte L. Prenatal diagnosis of congenital renal and urinary tract malformations. Facts Views Vis Obgyn 2011;3(3):165–74.
[Pubmed]
20.
Chudleigh T. Mild pyelectasis. Prenat Diagn 2001;21(11):936–41.
[Pubmed]
21.
Williams H. Renal revision: From lobulation to duplication—what is normal? Arch Dis Child Educ Pract Ed 2007;92(5):ep152–8. [CrossRef]
[Pubmed]
22.
Houat AP, Guimarães CTS, Takahashi MS, et al. Congenital anomalies of the upper urinary tract: A comprehensive review. Radiographics 2021;41(2):462–86. [CrossRef]
[Pubmed]
23.
Thotakura R, Anjum F. Hydronephrosis and Hydroureter. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
[Pubmed]
24.
Farrugia MK, Hitchcock R, Radford A, et al. British Association of Paediatric Urologists consensus statement on the management of the primary obstructive megaureter. J Pediatr Urol 2014;10(1):26–33. [CrossRef]
[Pubmed]
25.
Society for Maternal-Fetal Medicine (SMFM); Osmundson SS. Hydroureter. Am J Obstet Gynecol 2021;225(5):B16–7. [CrossRef]
[Pubmed]
26.
Has R, Sarac Sivrikoz T. Prenatal diagnosis and findings in ureteropelvic junction type hydronephrosis. Front Pediatr 2020;8:492. [CrossRef]
[Pubmed]
27.
Al Aaraj MS, Badreldin AM. Ureteropelvic Junction Obstruction. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
[Pubmed]
28.
Lissauer D, Morris RK, Kilby MD. Fetal lower urinary tract obstruction. Semin Fetal Neonatal Med 2007;12(6):464–70. [CrossRef]
[Pubmed]
29.
Kaefer M, Peters CA, Retik AB, Benacerraf BB. Increased renal echogenicity: A sonographic sign for differentiating between obstructive and nonobstructive etiologies of in utero bladder distension. J Urol 1997;158(3 Pt 2):1026–9.
[Pubmed]
30.
Robyr R, Benachi A, Daikha-Dahmane F, Martinovich J, Dumez Y, Ville Y. Correlation between ultrasound and anatomical findings in fetuses with lower urinary tract obstruction in the first half of pregnancy. Ultrasound Obstet Gynecol 2005;25(5):478–82. [CrossRef]
[Pubmed]
31.
Anumba DO, Scott JE, Plant ND, Robson SC. Diagnosis and outcome of fetal lower urinary tract obstruction in the northern region of England. Prenat Diagn 2005;25(1):7–13. [CrossRef]
[Pubmed]
32.
Chevalier RL. Congenital urinary tract obstruction: The long view. Adv Chronic Kidney Dis 2015;22(4):312–9. [CrossRef]
[Pubmed]
33.
Biard JM, Johnson MP, Carr MC, et al. Long-term outcomes in children treated by prenatal vesicoamniotic shunting for lower urinary tract obstruction. Obstet Gynecol 2005;106(3):503–8. [CrossRef]
[Pubmed]
34.
Scala C, McDonnell S, Murphy F, et al. Diagnostic accuracy of midtrimester antenatal ultrasound for multicystic dysplastic kidneys. Ultrasound Obstet Gynecol 2017;50(4):464–9. [CrossRef]
[Pubmed]
35.
Burgmaier K, Gimpel C, Schaefer F, Liebau M. Autosomal Recessive Polycystic Kidney Disease – PKHD1. In: Adam MP, Feldman J, Mirzaa GM, et al. editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2024.
[Pubmed]
36.
Goksu SY, Leslie SW, Khattar D. Renal Cystic Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
[Pubmed]
37.
Bergmann C. Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front Pediatr 2018;5:221. [CrossRef]
[Pubmed]
38.
Irfan A, O'Hare E, Jelin E. Fetal interventions for congenital renal anomalies. Transl Pediatr 2021;10(5):1506–17. [CrossRef]
[Pubmed]
39.
Shah HU, Ojili V. Multimodality imaging spectrum of complications of horseshoe kidney. Indian J Radiol Imaging 2017;27(2):133–40. [CrossRef]
[Pubmed]
40.
Vress D, Robertson M, Paoletti D. Fetal urinoma: A case report and review of the literature. Australas J Ultrasound Med 2015;18(1):38–40. [CrossRef]
[Pubmed]
41.
Farladansky-Gershnabel S, Gluska H, Meyer S, et al. Postnatal outcomes of fetuses with prenatal diagnosis of 6-9.9 mm pyelectasis. Children (Basel) 2023;10(2):407. [CrossRef]
[Pubmed]
42.
Choi YH, Cheon JE, Kim WS, Kim IO. Ultrasonography of hydronephrosis in the newborn: A practical review. Ultrasonography 2016;35(3):198–211. [CrossRef]
[Pubmed]
43.
Haeri S. Fetal Lower Urinary Tract Obstruction (LUTO): A practical review for providers. Matern Health Neonatol Perinatol 2015;1:26. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Acknowledgments
Radiological images were obtained from open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) with citation of the publication.
Author ContributionsKlara S Missling - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Kimberly S Sullivan - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Sarah Araji - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2024 Klara S Missling et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}